CASOS CLÍNICOS
Revista Peruana de Investigación en Salud, ISSN: 2616 - 6097 https://doi.org/10.35839/repis.17.1.1682
Epidermólisis bullosa en recién nacido
Bullous epidermolysis in newborn
Dana Molina-Piedra1, Yunier Cruz-Rodríguez1, Yailin Pérez-Díaz1
1Especialista de Primer grado en Medicina General Integral. Universidad de Ciencias Médicas, Villa Clara. Cuba
Corresponding author: E-mail:danamolina@infomed.sld.cu
Orcid ID: Molina-Piedra D.:https://orcid.org/0000-0001-9653-037X
Cruz-Rodríguez Y.: https://orcid.org/0000-0002-9843-3693
Pérez-Díaz Y.: https://orcid.org/0000-0002-7211-1215
Recibido: 12 de diciembre de 2022
Aprobado: 20 de febrero de 2023
En línea: 03 de marzo de 2023
Resumen
La epidermólisis bullosa es una enfermedad genética. Se manifiesta por la aparición de ampollas, úlceras posteriores a roces o traumatismos. Las lesiones pueden tener una localización cutánea y extra cutánea. Para realizar el diagnostico se debe tener presente los antecedentes heredofamiliares, la clínica, la microscopia y exámenes de laboratorio. Presentamos un caso de Epidermólisis Bullosa diagnosticado en recién nacido, femenino de 25 días en el Hospital Pediátrico Docente José Luis Miranda de Santa Clara. Se describen los hallazgos clínicos del caso. Se comenta acerca de los diferentes tipos de Epidermolisis y las formas de presentación por considerarse una entidad poco frecuente en la edad pediátrica.
Palabras clave: epidermólisis, bullosa, genética, edad pediátrica (fuente: DesCS-BIREME)
Abstract
Epidemolysis bullosa is a genetic disease. Clinical picture include blistering and ulcers after friction or trauma. Skin lesions may be cutaneous or extracutaneous. Family history, clinical findings, lab tests and microscopic examination are needed to make the diagnosis of the disease. A diagnosed case is presented in this paper, it is a 25 days female baby who was assisted at the Teaching Pediatric Hospital ″Jose Luis Miranda ″in Santa Clara. Clinical findings are described in the paper, as well as, comments about different types of Epidemolysis and their clinical manifestations, since this disease is not frequent in the pediatric age.
Keywords: epidemolysis, bullosa, genetics, pediatric age (source: MeSH-NLM)
Introducción
La epidermólisis bullosa o también conocida como piel de mariposa es una enfermedad autoinmune, de origen genético y afecta entre 15 y 17 nacidos por cada millón de habitantes. Se manifiesta por la presencia de ampollas, ulcerosas y heridas en la piel principalmente en mucosas, existen otras localizaciones extra cutáneas con alteraciones oculares ontogénicas gastrointestinales y musculo esqueléticas. La piel de los afectados con epidermólisis bullosa se caracteriza por ser frágil, débil, extremadamente sensible y muy vulnerable, tan delicada como el cristal; ya que al menor contacto físico se les desprende la piel(1-4). Pueden ser congénitas y adquiridas. Los tipos de epidermólisis bullosa son más graves en el período neonatal e incluso pueden ser mortales en los primeros meses y existen dos formas en las que la enfermedad se puede heredar: La herencia dominante, uno de los padres es portador del gen dominante por lo que tiene un 50 % de la descendencia con la enfermedad y el otro 50 % con genes normales. La herencia recesiva, en la que ambos padres son portadores de la enfermedad con un 25 % de probabilidades de procrear un hijo con genes normales, un 50 % que sea portador de la enfermedad y otro 25 % que padezca la afección(1, 2).
Existe alrededor de treinta subtipos de Epidermolisis Bullosa los cuales suelen agruparse en: El tipo simple: su localización es en epidermis, sobre todo en las manos y los pies, son muy dolorosas y suelen tener buena cicatrización. Es la más frecuente y menos letal, aparece al nacer. El tipo juntural se localizan entre la epidermis y la dermis, puede afectar la mucosa, esta variedad aparece con menor frecuencia. El tipo distrófica: se localizan las ampollas en la dermis, es la más frecuente, de mayor severidad y puede dejar secuelas produciendo deformidades en miembros superiores e inferiores. El síndrome de Kindler es una variedad donde ocurre fusión de varias capas, aparece en la infancia temprana y existe foto sensibilidad dándole un aspecto a la piel que varía de persona a persona(5, 6, 7,8).
Existen asociaciones civiles en todo el mundo llamada DEBRA (Dystrophic Epidermolysis Bullosa Research Association) con la misión de apoyar y ayudar a familiares y pacientes con Epidermolisis Bullosa, esta se ha extendido a 32 países del mundo(8).
Presentación del caso
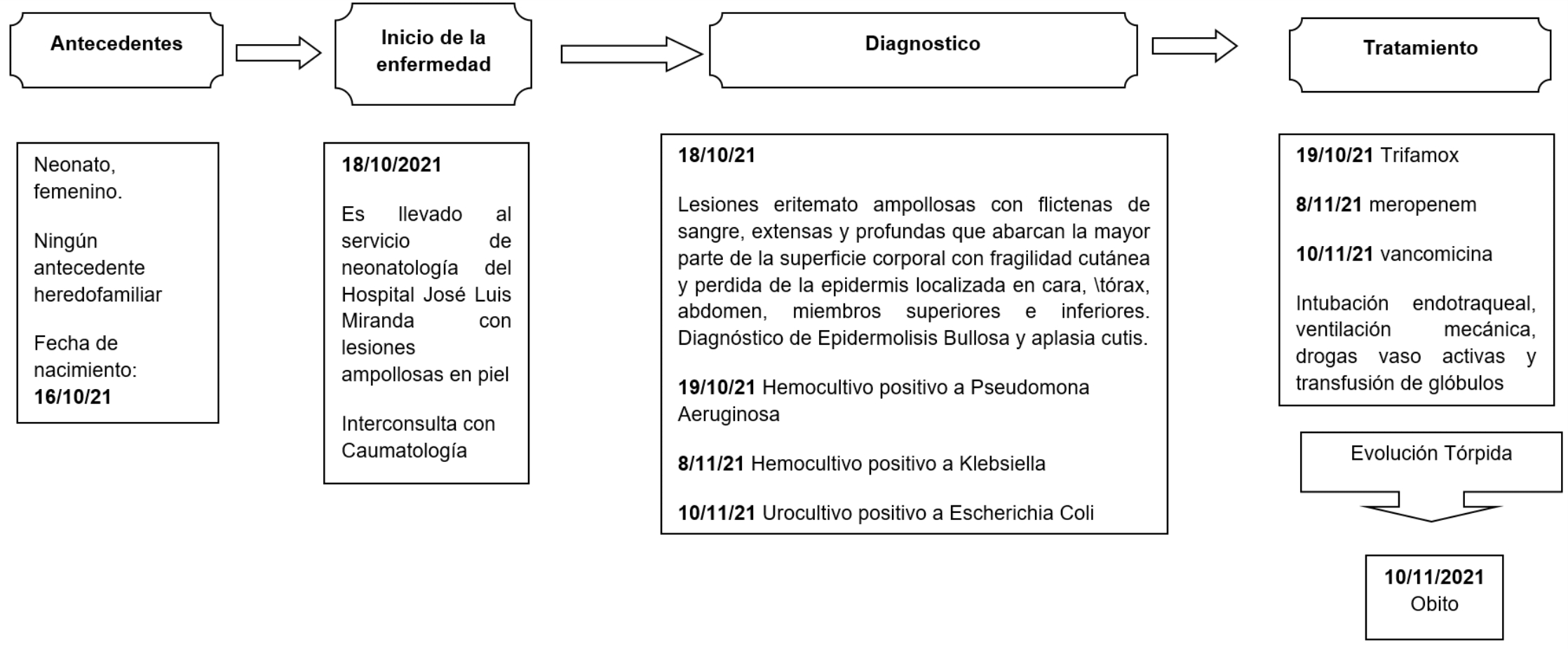
Neonato a término, de madre primigesta de 17 años con antecedentes obstétricos de gesta 1 parto 0 aborto 0, con edad gestacional de 39 semanas y antecedentes personales de hipertensión arterial crónica ,anemia ligera y vaginitis que fueron tratadas durante el embarazo, parto eutócico y apgar 8/9 con peso al nacer 3000 gramos presentando lesiones en la piel al nacimiento fue trasladado al hospital pediátrico José Luis Miranda a la sala de neonatología caracterizándose por lesiones eritemato ampollosas extensas y profundas que abarcan la mayor parte de la superficie corporal (65 %), con fragilidad cutánea y pérdida de la epidermis (Imagen: 1A, 1B, 2A, 2B, 3A) localizadas en cara, tórax, abdomen, miembros superiores e inferiores.
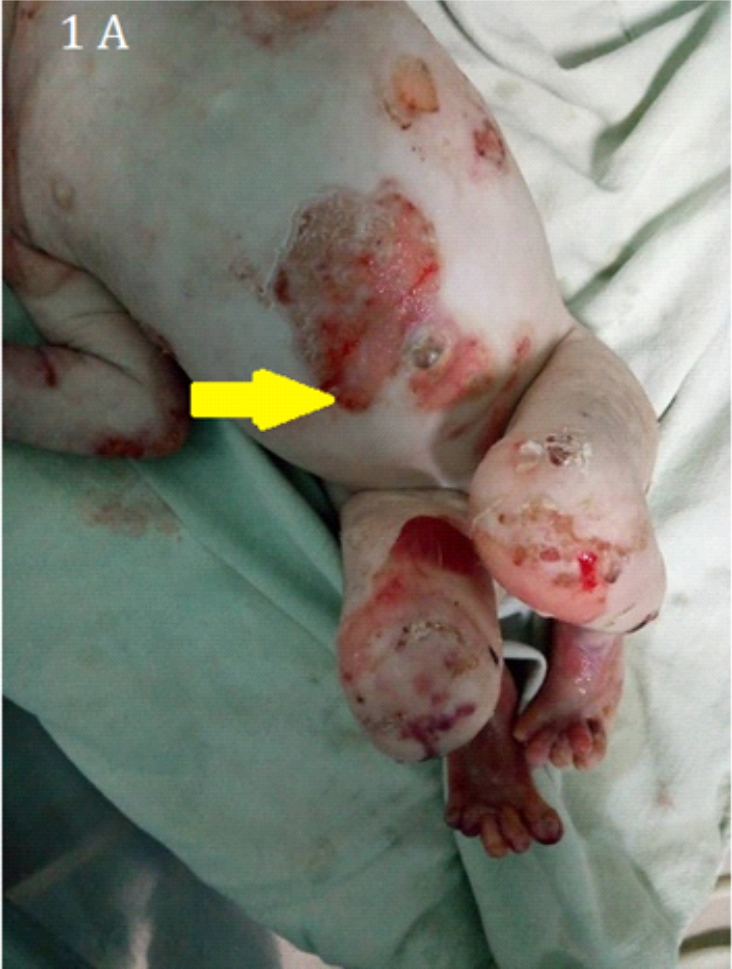
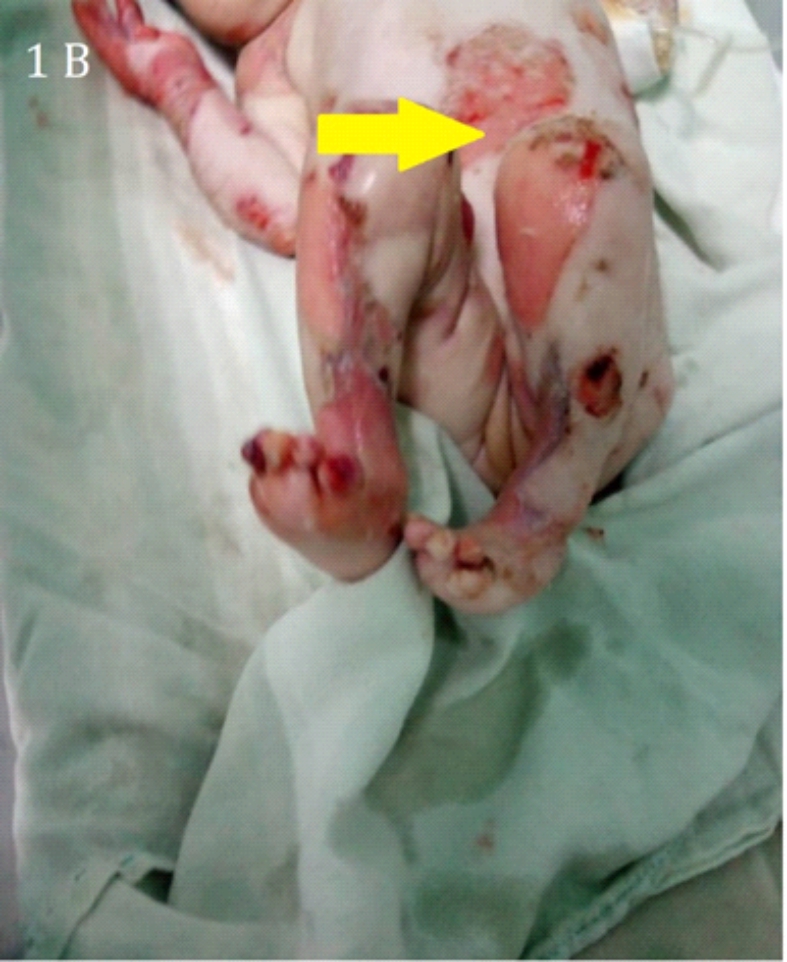
Imagen 1.Se observa en 1A una la piel frágil, sensible y dolorosa con aspecto de quemaduras localizadas en abdomen y en 1B se observa heridas en miembros inferiores al romper las ampollas
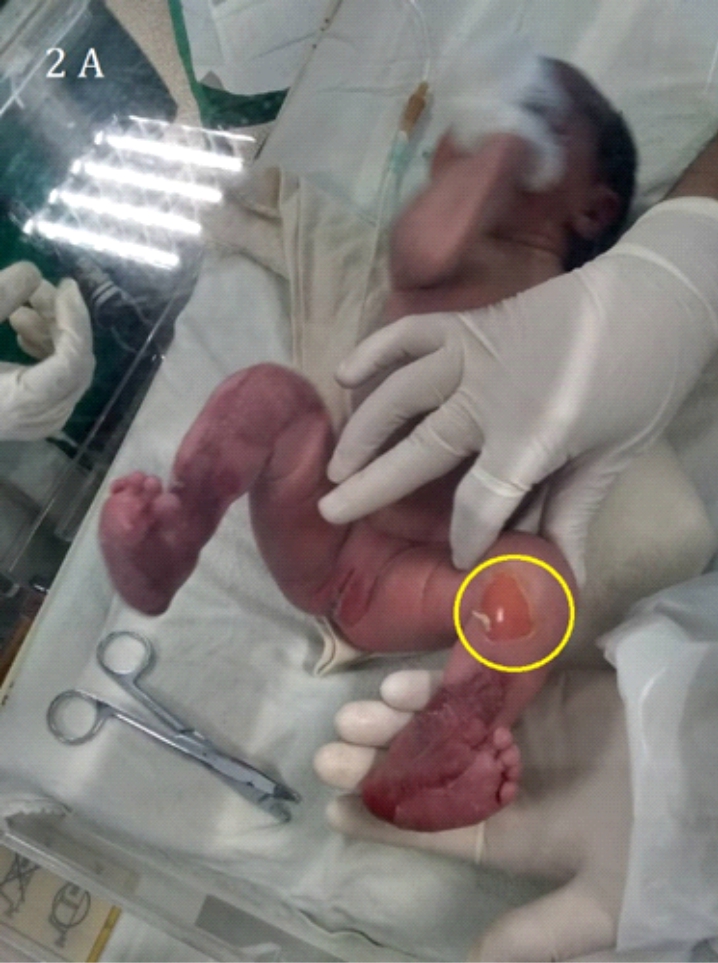
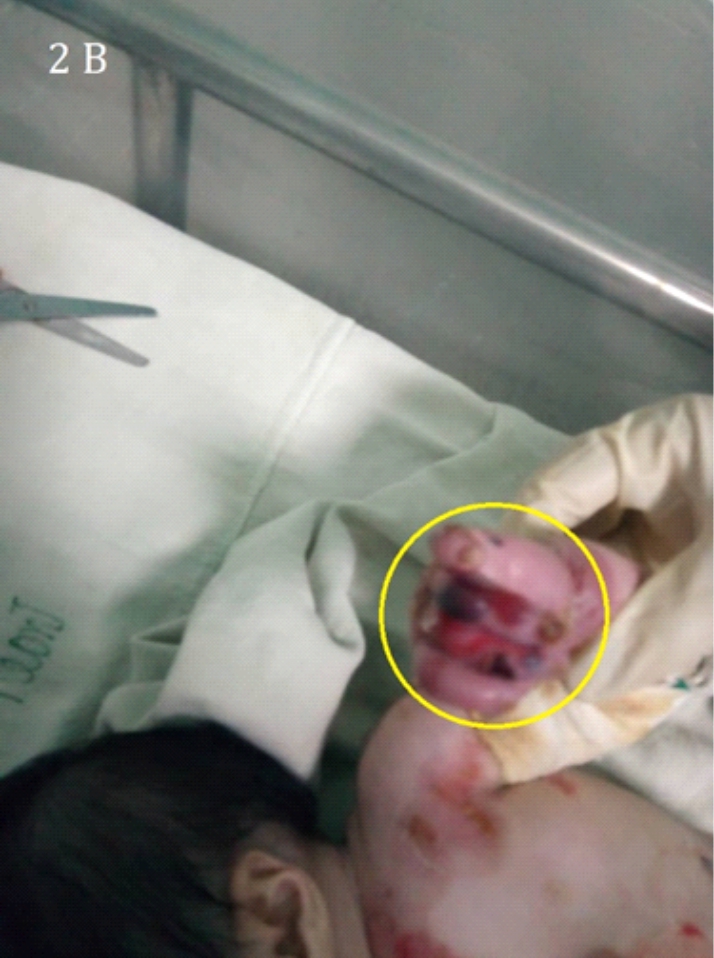
Imagen 2. Epidermólisis bullosa con lesiones necróticas ubicadas en miembros superiores 2B y ampollosas en miembros inferiores 2A
Imagen 3.Muestra la extensión de las lesiones con compromiso en cara, tórax, abdomen y miembros superiores e inferiores donde fue necesario el uso de vendajes húmedos
Según las evidencias anteriores se solicitó evaluación con Caumatología y se realizó el diagnostico de Epidermolisis Bullosa y Aplasia Cutis. Se solicitaron exámenes de laboratorio de los cuales se obtuvieron los siguientes resultados, Hemocultivo positivo a Pseudomona Aeruginosa, urocultivo positivo a Escherichia Coli con alta sensibilidad al meropenem. Recibió tratamiento antibiótico durante 10 días. Al aparecer nuevas lesiones y constatar empeoramiento del paciente se realizan nuevos complementarios.
El nuevo hemocultivo realizado muestra positivo a la Klebsiella asociándose vancomicina al tratamiento antimicrobiano. Se colocó catéter epicutáneo, el paciente comenzó a presentar bradicardia extrema cianosis palmo plantar, bradipnea con disociación térmica y palidez marcada. Seguidamente se realizó intubación endotraqueal y ventilación mecánica apoyada de drogas vaso activas, simultáneamente se administró transfusión de glóbulos. Cabe mencionar que a pesar de la terapéutica empleada la evolución fue tórpida lo que conllevó al óbito.
De modo que se descartaron enfermedades como pénfigo neonatal, herpes gestacional sobre todo porque no existen antecedentes familiares de Epidermolisis Bullosa.
Se cumplió los principios éticos contenidos en la Declaración de Helsinki, además contamos con el consentimiento informado de la familia del paciente.
La evolución clínica de nuestro paciente desde el inicio de su atención en el Hospital diagnóstico y tratamiento se describe a continuación (Figura 1).
Figura 1. Evolución clínica del paciente(1)
Discusión
La epidermólisis ampollosa es una entidad genética, hereditaria, crónica, compleja. Su diagnóstico y manifestaciones clínicas son poco conocidas. Es una enfermedad con baja prevalencia a nivel mundial. La Epidermolisis Bullosa simple se presenta con una mayor incidencia en países como Noruega, Escocia y norte de Irlanda, sin embargo, la Distrofia es más frecuente en los países del norte de Europa, mientras que la Juntural en Estados Unidos aparece en dos recién nascidos por millón de habitantes(1,2). En Costa rica se crea la asociación DEBRA latinoamericana representada por varios países como: México, Chile, Argentina y Brasil. La asociación DEBRA UK es una de las organizaciones mejores enfocadas en la investigación, apoyo, y soporte a los pacientes con Epidermolisis Bullosa. Según datos estadísticos hasta el 2019 se estima que en Cuba viven 200 personas con Epidermolisis Bullosa(3, 4, 5,6).
Para realizar el diagnostico de esta enfermedad en el neonato se deben tener en cuenta los antecedentes heredofamiliares y determinar el tipo de herencia autosómica dominante o autosómica recesiva, para descartar otras patologías, debemos usar el método clínico, confeccionando una historia clínica detallada, examen físico, describiendo las lesiones y técnicas histopatológicas. Se plantea que para llegar a realizar un diagnóstico integral en un paciente con Epidermolisis Bullosa se debe emplear el esquema en piel de cebolla realizándose de la siguiente manera: se determina el tipo mayor de Epidermolisis bullosa, fenotipo, modo de transmisión, sitio estructural de separación y hallazgos asociados, proteína involucrada, gen y tipo de mutación, así como la mutación especifica. La Epidermolisis Bullosa adquirida es infrecuente en los niños(7).
Los hallazgos cutáneos (ampollas) en los diferentes tipos de Epidermolisis Bullosa ocurren como consecuencia de la fragilidad cutánea. La gravedad de la enfermedad varía de acuerdo con las manifestaciones clínicas, desde la aparición de algunas ampollas que afectan las manos y los pies (Imagen: 1A,1B, 2A,2B y 3) hasta la muerte, la presencia de ampollas y cicatrización anormal en la piel y las mucosas contribuye a agravar la enfermedad. La manifestación extra cutánea más frecuente es la presencia de lesiones en la mucosa oral. La infección sobre añadida por estafilococos áureas puede llevar al paciente a un cuadro de septicemia fulminante (9, 10).
Aunado a esto es necesario resaltar las complicaciones que se derivan de la enfermedad al generalizarse las lesiones como se muestra en la figura 3, provocando deshidratación, desnutrición, en consecuencia, la muerte del bebé (11).
Estudios de otros autores mostraron que el estado nutricional que prevalece es la desnutrición en sus diferentes grados además de que predominar las afecciones musculo esqueléticas y de las mucosas (11,12).
Nuestro paciente con Epidermolisis Bullosa presentaba ampollas con costras figura 2 A y 2B sin embargo en la literatura consultada también se reportan; lesiones en la piel como quistes de milia, cicatrices y trastornos de la pigmentación (12).
Los padres deben ser capacitados en cuanto al cuidado y protección de la piel extremadamente vulnerable y mantener las medidas higiénicas sanitarias. En el caso que estamos presentando estos aspectos no se lograron cumplir por la hospitalización temprana de la paciente y la evolución desfavorable del paciente.
Conclusión
Aunque es una enfermedad rara y poco frecuente, la Epidermolisis Bullosa se debe conocer para poder realizar un diagnóstico temprano e iniciar la terapéutica oportunamente y de mayor eficacia para evitar las complicaciones. Principalmente debe estar orientado a evitar los traumas mecánicos y la cura de las lesiones evitando su crecimiento y propagación.
Contribución de los autores
1. Concibió la idea del manuscrito y análisis del estudio: Dana Molina Piedra
2. Escribió el primer borrador del artículo: Dana Molina Piedra
3. Metodología y Recolección de datos: Dana Molina Piedra, Yailin Pérez Díaz y Yunier Cruz Rodríguez
4. Realizó la edición crítica del artículo: Dana Molina Piedra, Yailin Pérez Díaz
5. Obtención de las fotografías: Dana Molina Piedra y Yunier Cruz Rodríguez
6. Aceptó el contenido final del artículo y aprobación de la versión final para publicación: Dana Molina Piedra, Yailin Pérez Díaz y Yunier Cruz
Referencias bibliográficas
1. Vázquez NMA, Santiesteban ARE, Ferrer MYI. Epidermólisis ampollosa o bullosa congénita. Actualización clínica. Rev. Finlay [Internet]. 2021 [citado el 16 mayo del 2022];11(1):74-79. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S2221-24342021000100074&lng=es.
2. Vidal G, Carrau F, Lizárraga M, Álvarez M. Epidermólisis ampollar: a propósito de un caso clínico. Arch. Pediatr. Urug. [Internet]. 2018 [citado el 16 mayo del 2022]; 89(6):382-388. Disponible en: http://www.scielo.edu.uy/scielo.php?script=sci_arttext&pid=S1688-12492018000700382
3. Revuelta ML, Ruíz RD, Guerra VD, Bravo PE. Epidermólisis bullosa. Presentación de un caso. Medisur [Internet]. 2016 [citado el 9 mayo del 2023]; 14(6). Disponible en: https://medisur.sld.cu/index.php/medisur/article/view/3175
4. Torres-Iberico, Rosario et al. Epidermólisis bullosa en el Perú: estudio clínico y epidemiológico de pacientes atendidos en un hospital pediátrico de referencia nacional, 1993-2015. Rev. Peru. med.exp. salud pública. 2017;34(2):201-208 doi: 10.17843/rpmesp.2017.342.2484
5. Del RC, Smith OY, González JAL, González DA, Arcis dRA, Fernández LY. Epidermólisis bullosa: piel de mariposa. A propósito de un caso. Rev.Med.Electrón. [Internet]. 2017 [citado el 16 mayo del 2022]; 39(3): 552-560. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1684-18242017000300013&lng=es
6. Dela-Rosa J, Zamora-Fung R, Vázquez-Gutiérrez G, López-Wilson A. Epidermólisis ampollosa, reporte de un caso. Universidad Médica Pinareña [Internet]. 2020 [citado 9 febrero 2023]; 17 (2) Disponible en: https://revgaleno.sld.cu/index.php/ump/article/view/529
7. Clavería CA, Rodríguez GK, Peña SM. Características clínicas, genéticas y epidemiológicas de la Epidermolisis bullosa y su repercusión en la cavidad bucal. Medisan [Internet]. 2015 [citado el 24 mayo 2022];19(8):995-1005. Disponible en: http://scielo.sld.cu/scielo.php?script=sciarttext&pid=S1029-30192015000800010&Ing=es.
8. Hernández SR, Morales MM, Castro RJA. Rehabilitación domiciliaria de la epidermólisis bullosa. Medicentro Electrónica [Internet]. 2021 [citado el 24 mayo 2022];25(1):126-136 Disponible en: http://scielo.sld.cu/scielo.php?script=sciarttext&pid=S1029-3043202110001001&Ing=es
9. Balleste LI, Campo GA, de los Reyes DR. Epidermolisis Bullosa a propósito de un caso. Rev Cubana Pedriatr [Internet].2008 [citado el 7 diciembre 2021];80(1) Disponible en: http://scielo.sld.cu/scielo.php?script=sciarttext&pid=S0034-75312008000100014&Ing=es.
10. Maldonado CG, Duran KC, Orozco CL, et al. Epidermolisis ampollosa nuevos conceptos clínicos moleculares para clasificación y diagnóstico. Artículo de revisión Dermatología Cosmética, médica y Quirúrgica [Internet]. 2016 [citado el 7 diciembre 2022]; 14(4)289-298. Disponible en: https://www.medigraphic.com/cgi-bin/new/resumen.cgi?IDARTICULO=70267
11. Araiza-Atanacio MI, Gris-Calvo J, Piña-Ramírez MJ, Cadena-León JF, de la Teja-Ángeles E, Varón-Munar D, et al. Epidermólisis ampollosa en niños: un estudio retrospectivo en un hospital de referencia. Rev. Med Inst. Mex Seguro Soc[Internet]. 2020[citado el 7 diciembre 2022]; 58(5):583-592. Disponible en: https://www.redalyc.org/journal/4577/457768466007/html/
12. Estrada PJE, Caro NII, Tibaduiza MYA, Sánchez SZM. Epidermólisis bullosa: presentación de un caso. Rev. Med. 2022; 29(2):121-6. doi.org/10.18359/rmed.5612
Esta obra está bajo una Licencia Creative Commons