Revista Peruana de Investigación en Salud, ISSN: 2616 - 6097 https://doi.org/10.35839/repis.5.3.1014
Caracterización clínica genética de pacientes con ataxias hereditarias en el estado de Portuguesa- Venezuela
Genetic clinical characterization of patients with hereditary ataxias in the state of Portuguesa-Venezuela
Daelys Castro-Montesino1,2,a, María B. Iglesias Rojas1,3,b, Omar Ramos-Fernández1,2,c, Adrián A. Rojas-Concepción1,2,d
1Universidad Popular Autónoma del Estado de Puebla, México
2Policlínico Universitario “Dr. Modesto Gómez Rubio”, Pinar del Río, Cuba
3Centro Provincial de Genética Médica, Pinar del Río, Cuba
Correspondencia: E-mail:daelyscm@infomed.sld.cu
Orcid ID: ahttps://orcid.org/0000-0001-8292-436X, bhttps://orcid.org/0000-0003-4816-9027, chttps://orcid.org/0000-0003-2351-0057, dhttps://orcid.org/0000-0003-3019-1453
Recibido el 21 de marzo de 2021
Aceptado para publicación: 30 de mayo de 2021
Resumen
Introducción: las ataxias hereditarias son enfermedades neurodegenerativas que provocan deterioro funcional. Como consecuencia de su carácter progresivo conllevan a la discapacidad en los individuos afectados con una severa afectación psicológica en sus familias. Objetivo: caracterizar clínico y genéticamente los pacientes con ataxias hereditarias en el estado Portuguesa- Venezuela en el periodo 2013-2015. Métodos: se realizó un estudio observacional, descriptivo de corte transversal en pacientes con diagnóstico de ataxia hereditaria. El universo estuvo constituido por 33 pacientes diagnosticados en el periodo de estudio, se trabajó con todos ellos. Se realizó la revisión de las historias de salud individual y familiar. Los datos obtenidos fueron procesados mediante programa estadístico SPSS. Resultados: existió predominio del grupo de edad de 25-29 años (18.18 %) y del sexo masculino (51.5%); así como de las ataxias espinocerebelosas autosómicas dominantes del adulto (SCA1 y la SCA2) (82 %). Los pacientes en etapa 3 de la enfermedad fueron los más predominantes (40 %). Predominó el tipo de herencia autosómica dominante (69 %) y la vía de herencia paterna (63 %). Conclusiones: la genética comunitaria y el asesoramiento genético contribuyeron con alternativas terapéuticas para mejorar la calidad de vida. Además, quedaron identificadas las poblaciones de riesgo para trazar acciones preventivas y reducir la recurrencia de las afecciones hereditarias.
Palabras clave: ataxias hereditarias, discapacidad, asesoramiento genético.
Abstract
Introduction: hereditary ataxias are neurodegenerative diseases that cause functional deterioration. As a consequence of their progressive nature, they lead to disability in affected individuals with severe psychological damage in their families. Objective: to characterize clinically and genetically the patients with hereditary ataxias in the Portuguese-Venezuela state in the period 2013-2015. Methods: an observational, descriptive cross-sectional study was carried out in patients with a diagnosis of hereditary ataxia. The universe consisted of 33 patients diagnosed in the study period, we worked with all of them. The individual and family health histories were reviewed. The data obtained were processed using the SPSS statistical program. Results: there was a predominance of the age group 25-29 years (18.18%) and the male sex (51.5%); as well as adult autosomal dominant spinocerebellar ataxias (SCA1 and SCA2) (82%). Stage 3 disease patients were the most prevalent (40%). The autosomal dominant type of inheritance predominated (69%) and the paternal inheritance path (63%). Conclusions: community genetics and genetic counseling contributed with therapeutic alternatives to improve the quality of life. In addition, the populations at risk were identified to outline preventive actions and reduce the recurrence of hereditary conditions.
Keyword: hereditary ataxias, disability, genetic counseling.
Introducción
Las ataxias espinocerebelosas autosómico dominantes (ADCA por sus siglas en inglés) constituyen un grupo clínica y genéticamente heterogéneo de enfermedades neurodegenerativas, caracterizadas por una ataxia progresiva variablemente asociada a otros signos neurológicos y causada por una degeneración gradual del cerebelo y del tallo cerebral. (1-5)
Según la edad de inicio de las ataxias hereditarias, estas suelen clasificarse en dos grandes grupos, las de inicio temprano y las tardías. Dentro del primero, se encuentran aquellas que se heredan con un patrón de herencia autosómico recesivo, asociadas o no a trastornos metabólicos. Las ataxias recesivas no asociadas a trastornos metabólicos definidos, son progresivas y entre ellas se encuentran las ataxias de Friedreich, por déficit de vitamina E, telangiectásica y otros síndromes que cursan con trastornos en la reparación del ADN, con apraxia oculomotora, espástica autosómica recesiva y mioclónica recesiva. (6-10)
Las ataxias hereditarias de inicio tardío o del adulto, generalmente se heredan con un patrón de herencia autosómico dominante. Estas se caracterizan por la degeneración aislada o predominantemente combinada del cerebelo, la médula espinal y sus vías de conexión, por lo que tradicionalmente se le conoce como Ataxias Espinocerebelosas (SCA, del inglés Spinocerebellar Ataxia). Dentro de las ataxias con patrón de herencia autosómico dominante se encuentran las SCA, los Síndromes Atáxicos, las Ataxias Episódicas y otros Síndromes Dominantes que cursan con Ataxia. (7-12)
La heterogeneidad genética de las Ataxias Autosómicas Dominantes sugiere que por lo menos faltan por identificar el 30% de la etiología molecular de las mismas. (11,13,15)
En algunas ocasiones, determinados trinucleótidos se encuentran repetidos en un número superior al rango normal y este hecho va asociado a la presen-cia de alguna patología generalmente neurológica y/o neuromuscular. Este tipo de alteración se conoce como mutación dinámica por presentar una serie de características que la hacen ser diferente a las clásicas mutaciones mendelianas que son estáticas. (12)
Las Ataxias SCA1, SCA2, SCA3, SCA6, SCA7, SCA17, la Atrofia dentato-rubro-luysiana, la atrofia espinobulbar y la enfermedad de Huntington, se encuentran dentro del grupo de las enfermedades causadas por expansiones del trinucleotido CAG, denominados también enfermedades poliglutamínicas por tener todos ellos un punto en común: la expansión de un triplete CAG que codifica para una glutamina. (11-16)
Los estudios de prevalencia en las ataxias hereditarias autosómicas dominantes son poco frecuentes a nivel internacional. Sin embargo, se ha estimado que el subtipo más común a nivel internacional es la SCA3, la cual representa un poco más del 30% de todas las ataxias hereditarias autosómicas dominantes. Las otras formas moleculares con valores significativos de prevalencia son la SCA2, SCA6, SCA7 y SCA8. (8,10,14,16,17)
El asesoramiento genético es el proceso de comunicación no dirigido que el especialista mantiene con una persona en relación al padecimiento, evolución o transmisión de una enfermedad de origen genético. La persona que solicita el asesoramiento genético puede estar afectada por la enfermedad (probando) o ser un familiar aparentemente sano del afectado (consultante). Durante el proceso de asesoramiento genético el profesional debe asegurarse de que al paciente y/o a la familia se le proporciona la información necesaria para: conocer y entender el diagnóstico realizado, su pronóstico y tratamiento si lo hubiere, el tipo de herencia, y el riesgo de recurrencia que supone, además conocer las alternativas disponibles para disminuir o eliminar el riesgo de recurrencia de la enfermedad, elegir una estrategia apropiada según el riesgo existente, los deseos de la familia y sus convicciones éticas o religiosas y adaptarse lo mejor posible a la nueva situación personal, familiar y sociolaboral.(19,20)
En Venezuela, se desconoce la prevalencia de las ataxias hereditarias, el número de familias afectadas y de sujetos en riesgo de enfermar, con la realización de esta investigación se aportará un cúmulo significativo de información que contribuirá a la caracterización epidemiológica, clínica, al entendimiento de la genética poblacional de estas patologías y a la implementación de estrategias de intervención tales como la rehabilitación física y cognitiva y al establecimiento del diagnóstico molecular. El estado portuguesa tiene un número significativo de familias con diagnóstico de enfermedades neurodegenerativas y ataxias hereditarias distribuidas en nueve municipios del estado, con un desconocimiento de la enfermedad y de la causa de la discapacidad.
La presente investigación se desarrolló con el objetivo de caracterizar clínico y genéticamente los pacientes con ataxias hereditarias en el estado Portuguesa- Venezuela en el periodo 2013-2015.
Métodos
Se realizó un estudio observacional, descriptivo de corte transversal en pacientes con diagnóstico de ataxia hereditaria en el estado Portuguesa de la República Bolivariana de Venezuela en el periodo 2013-2015. El universo se constituyó por 33 pacientes con diagnóstico de ataxia hereditaria en el periodo de estudio.
Se estableció una clasificación a partir del grado de validez de nuestros pacientes. (Anexo 1), así como la realización de algunos estudios como: examen neurológico y escala clínica.
Se les aplicó la Escala SARA (Scale for assessment and rating of ataxia) (Schmitz-Hübsch et al, 2006), para la estimación del daño cerebeloso, la cual permitió una evaluación de las alteraciones, incluyendo los siguientes aspectos: marcha, pararse (estancia de pie), sentarse, trastornos del lenguaje, seguimiento, movimientos alternativos de las manos, prueba talón – rodilla.
Las variables estudiadas fueron: sexo, edad, tipo de ataxia, etapas clínicas de la enfermedad y tipo de herencia.
Los datos obtenidos fueron depositados en una base de datos confeccionada al efecto y procesados en el paquete estadístico Statistical Packageforthe Social Sciences (SPSS) versión 21.0. Para describir el comportamiento de las variables se analizó de forma univariada mediante frecuencias absolutas y porcentajes. La investigación fue aprobada por el consejo científico y comité de ética. Se siguieron los principios éticos de la declaración de Helsinki.
Resultados
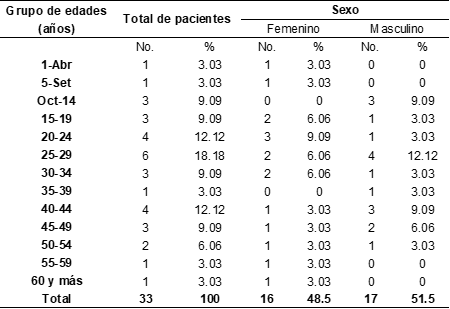
Existió predominio del grupo de edad de 25-29 años (18.18 %) y del sexo masculino (51.5%). (Tabla 1)
Tabla 1. Distribución según edad y sexo de pacientes con ataxia hereditaria del estado Portuguesa- Venezuela en el período 2013-2015
Se encontró predominio de las ataxias espino cerebelosas autosómicas dominantes del adulto (82 %), seguido de las ataxias de Friedrich (12 %) y las ataxias esporádicas (6 %).
Se evidenció que el 40 % de los pacientes del estudio se encontró en la etapa 3 de la enfermedad, seguido de la etapa 2 (33 %) y la etapa 1 (27 %)
Se encontró predominio del tipo de herencia autosómica dominante (69 %), seguido de la autosómica recesiva (19 %). En el 12 % no se precisó el tipo de herencia.
Resultaron superiores los que exhibían una herencia paterna (63 %), seguido de aquellos donde los dos padres eran portadores (25 %) y por último los que presentaron una vía de herencia materna (12 %).
Discusión
El estado Portuguesa ocupa el tercer lugar en el país con mayor prevalencia de discapacidad físicomotora y mixta por enfermedades genéticas como las ataxias hereditarias. Se detectaron en el primer estudio clínico-genético psicopedagógico y social 18 familias con diagnóstico de SCA. Estás familias se encuentran distribuidas en 9 municipios del estado: Unda, Guanare, Ospino, Guanarito, Papelón, Araure, Páez, Esteller, Turén.
Se han atendido 16 familias, se han detectado 6 casos nuevos, 14 fallecidos y se hanevaluado en consulta desde el punto de vista clínico-genético a 33 enfermos por equipos multidisciplinarios donde participan otros especialistas como neurólogo, defectólogo, fisiatra y genetista clínico según la disponibilidad de recursos humanos en las diferente sáreas de salud.
En la descripción de variables sociodemográficascomo edad y sexo, no se encontraron diferencias en relación a la afección y el sexo, por lo que no existe predominio de uno sobre el otro sino que afecta indistintamente. En relación a la edad, el mayor porciento de enfermos se encuentra en el rango de 20-59 años, lo que está en relación a las edades de debut de los primeros síntomas de la enfermedad y el fenómeno de anticipación que caracteriza la enfermedad. Resultados similares se observan en otros estudios realizados en Cuba (13,17).
Del total de enfermos evaluados, las ataxias espinocerebelosas autosómica dominante del adulto, del tipo: SCAs 2, 7, según características clínicas y signos neurológicos, son las más frecuentes en el presente estudio.
Esta sSCAs se encuentran dentro del grupo de las expansiones debidas a un incremento de trinucleótidos CAG, denominados también trastornos de las poliglutaminas. Desde el punto de vista epidemiológico son las más frecuentes en el continente americano, estudios demográficos señalan familias con SCAs tipo 3 en Brasil, SCAs tipo 2 en Cuba y SCAs tipo 7 en Venezuela, asícomo la ataxia de Friedreich es la ataxia heredada más frecuente en Estados Unidos. (3, 4, 7,11-13)
Conocer la historia genética familiar mediante el árbol genealógico como principal herramienta profesional en el campo de la genética médica es el primer paso para la identificación del riesgo genético, que en términos de genética comunitaria se dirige a la búsqueda de la predisposición para enfermedades de origen mendeliano y el riesgo genético reproductivo. La introducción de servicios de genética clínica en la comunidad; el asesoramiento genético permite realizar estrategias que vayan dirigidas a disminuir la recurrencia de discapacidades por estas causas, ofreciendo a la población una información adecuada de sus riesgos, acerca de la planificación de su descendencia para mejorar la calidad de vida y educación en general de las poblaciones vulnerables. (18-20)
El principal objetivo de las aplicaciones de la genética a la salud pública es la reducción del impacto de las enfermedades genéticas sobre la salud y el bienestar de los individuos mediante estrategias de prevención, así como reducir su frecuencia, contribuyendo a la modificación del acervo genético de nuestra especie, ayudando de manera pasiva actuando sobre familias individuales o de manera activa a través de políticas de salud que contribuyan a mejorar la calidad de vida de la población en las comunidades donde existe mayor prevalencia.(19,20)
Teniendo en cuenta que en las ataxias hereditarias autosómica dominante del adulto existe un fenómeno de anticipación es muy importante trazar estrategias de prevención y de asesoramiento a estas familias explicando la posibilidad de que estos descendientes sean personas presintomáticas.
La neurorrehabilitación y la vitaminoterapia en altasdosispermitenmejorar la calidad de vida de las personas con ataxias espinocerebelosas mejorando el estado cognitivo y los síntomas de forma general. Estudiosrealizados en Cuba demuestran beneficios del ejerciciofísico en pacientes con diagnóstico de Ataxias hereditarias en estadios-ligeros. (17-20)
Se encontró un predominio del grupo de edades de 25-29 años y del sexo masculino; las ataxias espino cerebelosas autosómicas dominantes del adulto fueron las más frecuentes. La mayoría de los pacientes se encontraron en la etapa 3 de la enfermedad. Predominaron el tipo de herencia autosómica dominante y la herencia paterna.
Contribución de los autores
DCM participó en la concepción y diseño de la investigación y la recolección de los datos. DCM, MBIRyORF participaron en el procesamiento estadístico de los datos. DCM, MBIRyAARC se encargaron de la redacción del artículo. Todos los autores revisaron y aprobaron el manuscrito y su versión final.
Conflicto de Interés
Los autores declaran que no existe ningún conflicto de intereses.
Referencias
1. SaffieAwad P, Vial Undurraga F, Chaná Cuevas P. Características clínicas de 63 pacientes con ataxia. Rev Med Chile. [Internet] 2018 [Citado 3Febrero 2020]; 146: [Aprox. 5 p.]. Disponibleen: https://scielo.conicyt.cl/pdf/rmc/v146n6/0034-9887-rmc-146-06-0702.pdf
2. Margarida de Castro A. Ataxia aguda en la edad pediátrica. Rev Clínica.[Internet]2017 [Citado 3 Febrero 2020]; 26: [Aprox. 9 p.]. Disponible en: https://doi.org/10.24197/cl.26.2017.29-38
3. Drumond MT, Prado M, Vasconcellos LF. Ataxia cerebelar idiopática de início tardio (ILOCA): un desafio diagnóstico. Rev Bras Neurol. [Internet] 2015 ene- mar [Citado 3Febrero 2020]; 51(1): [Aprox. 12 p.]. Disponible en: http://files.bvs.br/upload/S/0101-8469/2015/v51n1/a4730.pdf
4. Vieira de Rezende-Pinto WB, Pedroso JL, Sgobbi de Souza PV, Cristino de Albuquerque MV, Povoas Barsottini OG. Ataxias cerebelares não-progressivas e lesão cerebelar aguda prévia indeterminada: uma condição clínica misteriosa. Rev ArqNeuro-Psiquiatr. [Internet] 2015 oct[Citado 3Febrero 2020]; 73(10): [Aprox. 12 p.]. Disponibleen: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-282X2015001000823.http://dx.doi.org/10.1590/0004-282X20150119
5. Díaz de la Fe A. La rehabilitación en las ataxias. Rev Mex Neurociencias. [Internet] 2014 jul- ago [Citado 3Febrero 2020]; 15(4): [Aprox. 5 p.]. Disponible en: https://www.medigraphic.com/cgi-bin/new/resumen.cgi?IDARTICULO=51741
6. Cristino de Albuquerque MV, Pedroso JL, Braga Neto P, Povoas Barsottini OG. Variabilidade fenotípica e ataxia de início precocena ataxia spinocerebelar tipo 7: correlaçõescomoutras ataxias espinocerebelares. Rev Arq Neuro-Psiquiatr. [Internet] 2015 jan [Citado 3Febrero 2020]; 73(1): [Aprox. 5 p.]. Disponible en: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-282X2015000100018
7. Moro A, Munhoz RP, Arruda WO, Raskin S, Moscovich M, Teive AG. Ataxia espinocerebelar do tipo 3: subfenótipos em uma coorte de pacientes brasileiros. Rev ArqNeuro-Psiquiatr. [Internet] 2014 sep[Citado 3Febrero 2020]; 72(9): [Aprox. 9 p.]. Disponible en: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-282X2014000900659
8. TorralbasFitz SJ, Velázquez Pérez L, Torralbas Blázquez MJ, Velázquez González VA, Rodríguez Labrada R. Ataxia espinocerebelosa tipo 2 y síndrome de Ehlers-Danlos: a propósito de un caso. Rev Archivo Médico de Camaguey. [Internet] 2016 may-jun [Citado 3Febrero 2020]; 20(3): [Aprox. 5 p.]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1025-02552016000300013
9. Diaz Novo C, Gámez Rodríguez O, Montoya Pedrón A, VigilZulueta IA, Zamora Matamoros L. Descripción cinemática de la marcha en pacientes con ataxia espinocerebelosa de tipo 2. Rev Medisan. [Internet] 2016 dic [Citado 3Febrero 2020]; 20(12): [Aprox. 5 p.]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1029-30192016001200003
10. Gordo Gómez YM, Ramírez Guerra DM, Rodríguez Labrada R, Velázquez Pérez LC. Alteraciones respiratorias de la Ataxia Espino-cerebelosa Tipo 2: de las bases fisiopatológicas a su impacto en la neurorrehabilitación. Rev Correo Científico Médico. [Internet] 2018 ene- mar [Citado 3Febrero 2020]; 22(1): [Aprox. 7 p.]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_abstract&pid=S1560-43812018000100010&lng=es&nrm=iso
11. Martínez Guerrero J, Paz Gutiérrez J, Vega Gaxiola S.B. Ataxia espinocerebelosa tipo 2. Rev ArchNeurocien INNN. [Internet] 2016 ene- mar [Citado 3Febrero 2020]; 21(1): [Aprox. 11 p.]. Disponibleen: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=ES&Expert=98756
12. Rodríguez Labrada R. Ataxia espinocerebelosa tipo 2: estudio de los movimientos oculares sacádicos en familias portadoras de la Mutación SCA2. Tesis de Doctorado. Universidad de Ciencias Médicas de la Habana; 2015. Disponible en: http://eduniv.reduniv.edu.cu/index.php?page=13&id=472&db=1
13. Rojo Suárez N, Marín Iglesias MR. Ataxia espinocerebelosa hereditaria por déficit de vitamina E y la importancia del estudio genético. Rev Genética Médica y Genómica. [Internet] 2017 abr [Citado 3Febrero 2020]; 1(1): [Aprox. 4 p.]. Disponibleen: https://fedaes.org/ataxia-espinocerebelosa-hereditaria-deficit-vitamina-e-la-importancia-del-estudio-genetico/
14. Perdomo-Rebollo FG. Escala BARS en niños con ataxia. Rev Med Inst Mex Seguro Soc. [Internet] 2017 [Citado 3Febrero 2020]; 55(6): [Aprox. 4 p.]. Disponible en: https://www.medigraphic.com/pdfs/imss/im-2017/im176g.pdf
15. Velázquez-Palacio R, Rodríguez-Labrador R, Velázquez-Pérez L. Importancia del cinc en el sistema nervioso: la Ataxia Espinocerebelosa Tipo 2 como modelo. Rev Mexicana de Neurociencia. [Internet] 2017 [Citado 3Febrero 2020]; 18(3): [Aprox. 10 p.]. Disponible en: https://www.medigraphic.com/pdfs/revmexneu/rmn-2017/rmn173f.pdf
16. Velázquez Pérez L. Nueva era en las investiga-ciones e intervención sobre la ataxia espino cerebelosa tipo 2. Rev Correo Científico Médico. [Internet] 2015 oct- dic [Citado 3Febrero 2020]; 19(4): [Aprox. 9 p.]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1560-43812015000400001
17. Fernández Martínez E, Jorge Rodríguez JL, Rodríguez Pérez D, Crespo Moinelo M, Fernán-dez Paz J. La neurorrehabilitación como alternativa esencial en el abordaje terapéutico de las ataxias cerebelosas. Rev Cubana Salud Pública. [Internet] 2013 jul- sep [Citado 3Febrero 2020]; 39(3): [Aprox. 4 p.]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-34662013000300007
18. Restrepo CM. ¿Es el asesoramiento genético una práctica que estimula la eugenesia?. Rev Cienc Salud Bogotá. [Internet] 2018 ene- abr [Citado 3Febrero 2020]; 16(1): [Aprox. 2 p.]. Disponible en: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S1692-72732018000100007
19. Alonso Gordo JM. Las posibilidades del consejo genético en Atención Primaria. Rev ClínMed Fam. [Internet] 2014 [Citado 3Febrero 2020]; 7(2): [Aprox. 11 p.]. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1699-695X2014000200006
Esta obra está bajo una Licencia Creative Commons